There are two types of CE instruments for fragment analysis. However, only the ones with Sanger sequencing capability can match the requirements (e.g. ThermoFisher 3730xl). The comparisons are listed in the following table.
| Category | Instrument with Sanger sequencing capability | Instrument without Sanger sequencing capability |
|---|---|---|
| Example Instrument | ThermoFisher 3730xl etc. | AATI Fragment Analyzer |
| Mutation amplification technology | PCR with at least one fluorescent labeled primer | PCR without fluorescent labeled primers |
| Peak representation | Single-stranded or double-stranded fragments with fluorescent labeling dye at end | Double-stranded fragments with intercalating fluorescent dye |
| Can reach single bp resolution? | Yes | No |
| Depend on enzyme cleavage? | No | Yes |
| Protocol | Short: PCR – CE | Long: PCR – Heat/cool–Enzyme cleavage – CE |
| Mutation Efficiency/frequency estimation (comparing to NGS) | Comparable 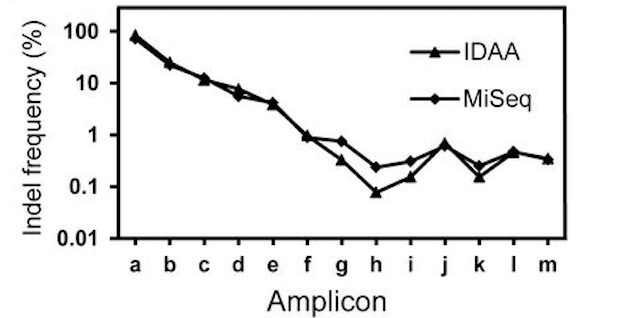 |
Underestimate
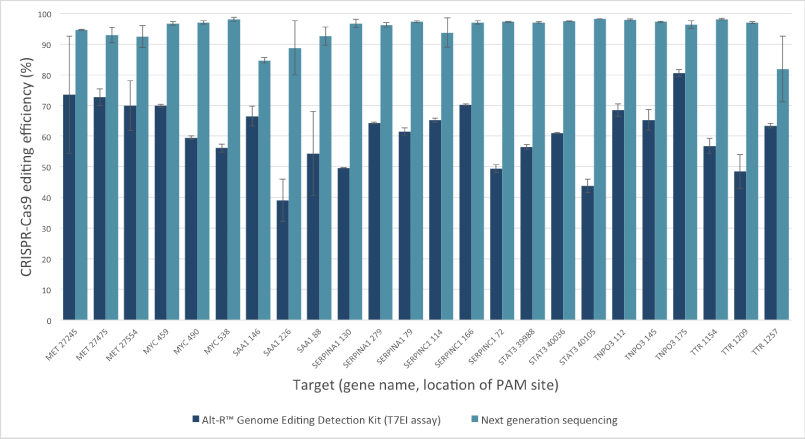
|
| High-throughput? | Yes | Yes |
| Quantitative analysis software | Primary: GeneMapper or Peak Scanner (free) Secondary: fragmentanalysis.com | PROSize® |
| Applied for more than 1kb fragment? | No | Yes |
| Price | Depend on users, less than 1-3$/sample for many core facilities or companies | Depend on users |
It is recommended to use Google Chrome, Mozilla Firefox and Safari.
Yes. You can do analysis with all kinds of sample name formats. However, the statistical analysis is only based on predefinded format, as described in the tutorial.
You need update your browser.
Yes. There is an icon at the top right of each chart, click it, you will see the options. ![]()
For the desktop version of Peak Scanner (v1 or v2), in the left navigation part, there is a link “New Size Standard” under the resources section. Click this link, then a window will pop up. Fill in the following contents:
Name: the name of the new standard Color: Choose Red if use ROX, Orange if use LIZ Description: it is optional
In the big window, put your size standard there, with comma separated for each size. And then click "Add Sizes >>" and “Save”. Now the new size standard will be listed in the drop down list of “Size Standard”.
The similar approach is available for cloud version of Peak Scanner.
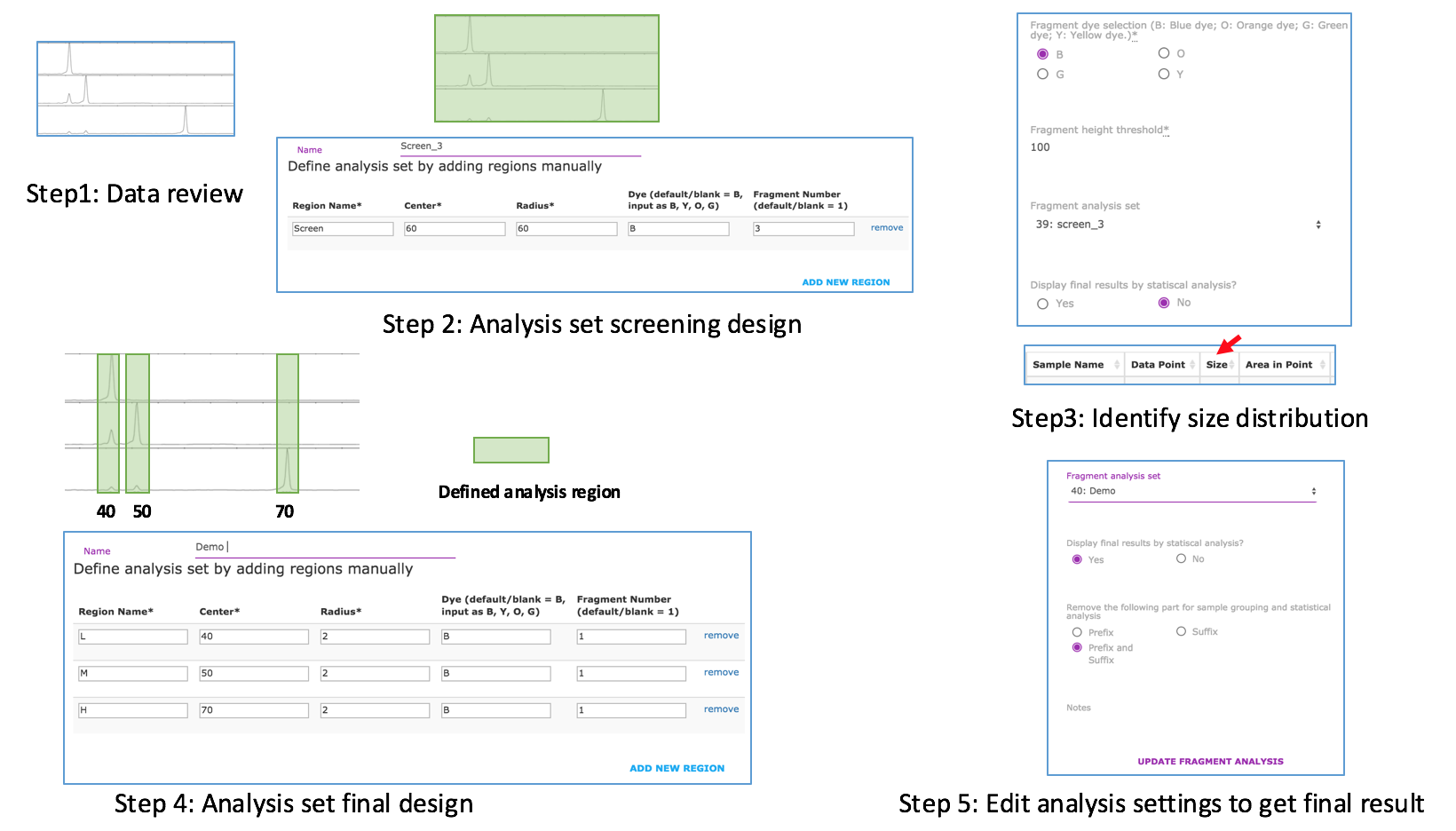
Steps for initial data analysis: